R and the nat libraries¶
NAVis is inspired by the excellent nat (NeuroAnatomy Toolbox) ecosystem by Greg Jefferis . While there is parity in major features between the navis ecosystem and the “natverse”, it can sometimes be useful to access R functions directly.
Fortunately, Python and R play very nicely together - see this brilliant blog post. Hence, NAVis has functions for converting from/to nat datatypes and basic wrappers for some of the most useful nat functions. This section will teach you the basics of how to use R and NAVis. But first, we have to make sure you are all set:
Setting up¶
Using R from within Python requires rpy2. rpy2 is not automatically installed alongside NAVis. That’s because it fails to install if R is not already present on your system. Here is what you need to do:
-
Install R.
You can either install just R or install it along with R Studio (recommended). -
Install rpy2
This should do the trick:pip3 install rpy2
Check out rpy2's documentation if you are running into issues. -
Install R packages.
NAVis has wrappers for the nat (NeuroAnatomy Toolbox) ecosystem by Greg Jefferis. Please make sure to install:- nat - core package for morphological analysis of neurons
- nat.nblast - morphological similarity
- elmr - bridging between EM and light level data
- rcatmaid - interface with CATMAID
- flycircuit - interface with the flycircuit database for fly neurons
- nat.templatebrains and nat.flybrains - bridging between different light level template brains
You are ready to go! On a fundamental level, you can now use every single R function from within Python - check out the rpy2 documentation.
Quickstart¶
import navis
navis.set_pbars(hide=True, jupyter=False)
from navis.interfaces import r
import matplotlib.pyplot as plt
from rpy2.robjects.packages import importr
import rpy2.robjects as robjects
Load nat as module
nat = importr('nat')
Run one of the nat examples in Python
# Get neurons (Drosophila Kenyon cells) shipped with nat
kcs20 = robjects.r('kcs20')
head = robjects.r('head')
head(kcs20)
| gene_name | Name | idid | ... | cluster | idx | type |
|---|---|---|---|---|---|---|
| 'FruM... | 'fru-... | 1024.000000 | ... | 9 | 156 | gamma |
| 'GadM... | 'Gad1... | 10616.000000 | ... | 70 | 1,519 | gamma |
| 'GadM... | 'Gad1... | 8399.000000 | ... | 57 | 1,132 | ab |
| 'GadM... | 'Gad1... | 10647.000000 | ... | 71 | 1,535 | apbp |
| 'FruM... | 'fru-... | 9758.000000 | ... | 64 | 1,331 | ab |
| 'FruM... | 'fru-... | 6182.000000 | ... | 44 | 795 | ab |
Convert the Kenyon cells to NAVis DotProps
kcs20_py = r.dotprops2py(kcs20)
kcs20_py.head()
| gene_name | Name | idid | soma_side | flipped | Driver | Gender | X | Y | Z | exemplar | cluster | idx | type | points | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | FruMARCM-M001205_seg002 | fru-M-500112 | 1024.0 | L | 0 | fru-Gal4 | M | 361.484872 | 95.044800 | 84.102594 | FruMARCM-M001205_seg002 | 9 | 156 | gamma | x y z x_v... |
| 1 | GadMARCM-F000122_seg001 | Gad1-F-900005 | 10616.0 | L | 0 | Gad1-Gal4 | F | 367.833167 | 105.867548 | 94.734459 | GadMARCM-F000122_seg001 | 70 | 1519 | gamma | x y z x_v... |
| 2 | GadMARCM-F000050_seg001 | Gad1-F-100010 | 8399.0 | R | 1 | Gad1-Gal4 | F | 382.827911 | 61.732132 | 97.280571 | GadMARCM-F000050_seg001 | 57 | 1132 | ab | x y z x_ve... |
| 3 | GadMARCM-F000142_seg002 | Gad1-F-300043 | 10647.0 | L | 0 | Gad1-Gal4 | F | 349.591724 | 78.189859 | 96.692797 | GadMARCM-F000142_seg002 | 71 | 1535 | apbp | x y z x_ve... |
| 4 | FruMARCM-F000270_seg001 | fru-F-400045 | 9758.0 | L | 0 | fru-Gal4 | F | 387.523611 | 114.803444 | 87.841563 | FruMARCM-F000270_seg001 | 64 | 1331 | ab | x y z x_v... |
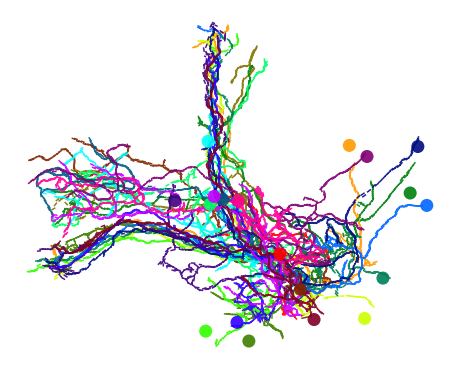
Plot them
fig, ax = navis.plot2d(kcs20_py, linewidth=1.5)
Data conversion¶
navis.interfaces.r provides functions to convert data from Python to R:
navis.interfaces.r.data2py()converts general data from R to Pythonnavis.interfaces.r.neuron2py()converts R neuron or neuronlist objects to Pythonnavis.TreeNeuronandnavis.NeuronList, respectivelynavis.interfaces.r.neuron2r()convertsnavis.TreeNeuronornavis.NeuronListto R neuron or neuronlist objectsnavis.interfaces.r.dotprops2py()converts R dotprop objects to pandas DataFrame that can be passed tonavis.plot.plot3d()
R NBLAST¶
NAVis has a native (i.e. pure Python) implementation of NBLAST (see this Tutorial) that matches the one in nat. While I recommend that you use the Python implementation, you can also use the nat interface to run the R NBLAST:
|
NBLAST using R's |
|
All-by-all NBLAST using R's |
|
Class holding NBLAST results and wrappers that allow easy plotting. |
All-by-all nblast¶
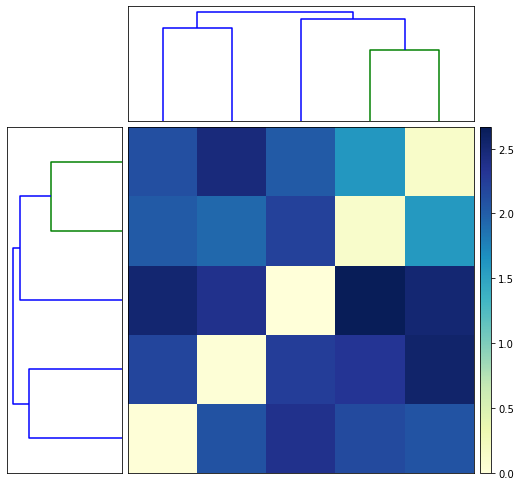
Comparing neurons which are already in the same brain space is straight forward:
nl = navis.example_neurons()
# Note that we are converting to microns as NBLAST is optimized for that resolution
nbl = r.nblast_allbyall(nl, micron_conversion=1/1000, resample=1)
fig = nbl.plot_matrix()
INFO : Use matplotlib.pyplot.show() to render figure. (navis)
NBLAST against databases¶
In this example we will nblast one of our example Drosophila neurons against the flycircuit database of single cell clones.
This is a two step process in which the neuron is first transformed from it’s original brain space (FAFB14) into the flycircuit template space (FCWB) and then blasted against the entire database of skeletonized flycircuit clones.
This requires you to have the R flycircuit and nat.templatebrains packages installed.
n = navis.example_neurons(1)
# Get the flycircuit database
datadir = robjects.r('getOption("flycircuit.datadir")')[0]
fc = robjects.r(f'read.neuronlistfh("{datadir}/dpscanon.rds")')
nbl = r.nblast(n, db=fc, xform='FAFB14->FCWB', resample=1, mirror=True)
INFO : Blasting neuron... (navis)
INFO : Blasting done in 67.3 seconds (navis)
Show the top results
nbl.results.head()
| gene_name | forward_score | reverse_score | mu_score | |
|---|---|---|---|---|
| 0 | DvGlutMARCM-F002430_seg001 | 0.614162 | 0.632155 | 0.623158 |
| 1 | FruMARCM-M002373_seg002 | 0.603885 | 0.681603 | 0.642744 |
| 2 | dTdc2MARCM-F000115_seg001 | 0.588686 | -0.498178 | 0.045254 |
| 3 | ChaMARCM-F000725_seg001 | 0.566431 | 0.676217 | 0.621324 |
| 4 | GadMARCM-F000709_seg001 | 0.565479 | 0.566549 | 0.566014 |
Plot the top 2 hits ontop of the query neuron (black).
fig = nbl.plot3d(hits=2, width=1000)